Proteins
Proteins are nitrogenous organic compounds used throughout the body. They play an important role in many critical functions of living organisms. Specifically, in the human body, they carry out various essential functions from structural cell support to enzymatic activity. Proteins are made up of amino acids which are organic compounds containing amine and carboxyl functional groups. They are formed when DNA goes through the process of transcription to form a messenger ribonucleic acid (mRNA) and this mRNA is then translated to synthesize proteins.
Proteins are nitrogenous organic compounds used throughout the body. They play an important role in many critical functions of living organisms. Specifically, in the human body, they carry out various essential functions from structural cell support to enzymatic activity. Proteins are made up of amino acids which are organic compounds containing amine and carboxyl functional groups. They are formed when DNA goes through the process of transcription to form a messenger ribonucleic acid (mRNA) and this mRNA is then translated to synthesize proteins.
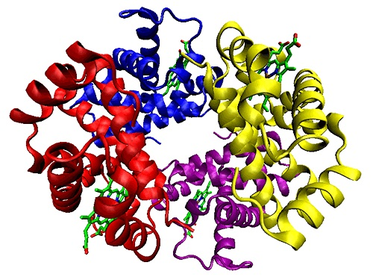
Ribbon diagram of the structure of hemoglobin (a protein)
Ligands
Ligands are substances that form complexes with biological molecules to serve a specific purpose. For example, heme, a ligand that contains an iron ion, binds to hemoglobin within red blood cells so they can successfully carry oxygen around the body. These ligands bind to protein at specific sites, allowing a biological complex to be created. In some cases, a slight mutation to a protein may cause its binding sites to be incorrectly configured, potentially resulting in important ligands not being able to bind to the protein.
Ligands are substances that form complexes with biological molecules to serve a specific purpose. For example, heme, a ligand that contains an iron ion, binds to hemoglobin within red blood cells so they can successfully carry oxygen around the body. These ligands bind to protein at specific sites, allowing a biological complex to be created. In some cases, a slight mutation to a protein may cause its binding sites to be incorrectly configured, potentially resulting in important ligands not being able to bind to the protein.
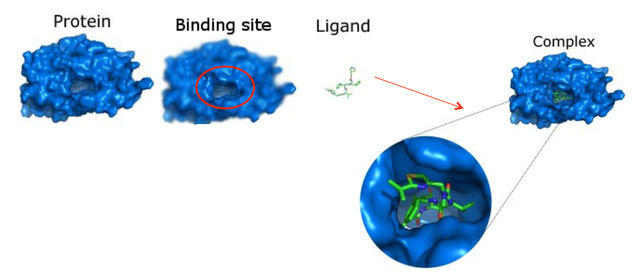
Potential Factors Used to Identify Protein Binding Sites
Geometric Factors
Concavities, such as pockets/cavities, most likely indicate a protein binding site. It is unlikely for a ligand to bind to the very exterior of a protein. On the other hand, ligands prefer to be surrounded by the protein, allowing a large surface of potential interaction between the protein and ligand. This also ensures that the complex formed by the protein and the ligand is relatively stable as opposed to the ligand breaking off from the complex.
Concavities, such as pockets/cavities, most likely indicate a protein binding site. It is unlikely for a ligand to bind to the very exterior of a protein. On the other hand, ligands prefer to be surrounded by the protein, allowing a large surface of potential interaction between the protein and ligand. This also ensures that the complex formed by the protein and the ligand is relatively stable as opposed to the ligand breaking off from the complex.
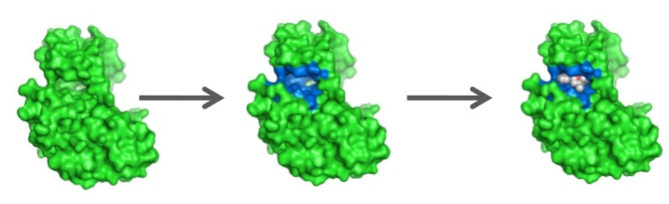
An example of a pocket within a protein that can house a ligand.
Conservation Information
Usually, residues that bind ligands are conserved. This means that the same sequence (amino acids for a residue) can be found in multiple species of the protein that are distantly related (orthologous sequences) or within a genome (paralogous sequences). Conservation information of the protein sequence can be extracted from multiple sequence alignments (MSAs). Then, it can be used to improve the prediction of structure-based approaches or used on its own to predict protein binding sites.
Usually, residues that bind ligands are conserved. This means that the same sequence (amino acids for a residue) can be found in multiple species of the protein that are distantly related (orthologous sequences) or within a genome (paralogous sequences). Conservation information of the protein sequence can be extracted from multiple sequence alignments (MSAs). Then, it can be used to improve the prediction of structure-based approaches or used on its own to predict protein binding sites.

A multiple sequence alignment of five mammalian histone H1 proteins is shown above. Residues that are conserved across all sequences are highlighted in grey. Below each position of the protein sequence alignment is a key:
- conserved sites (*) – same amino acid
- sites with conservative replacements (:) – very similar biochemical properties
- sites with semi-conservative replacements (.) – relatively similar
- sites with non-conservative replacements ( ) – radically different
Holo-structures versus Apo-structures
Having the holo-structure of a protein is usually preferred in drug design, when predicting protein binding sites and finding their shape/size. The holo-structure of a protein (conjugated protein) is the apo-protein combined with its prosthetic group (a ligand). Many enzymes may require cofactors or coenzymes in order to function fully. Such cofactors or coenzymes would be present within the holo-structure of the protein. Due to the induced fit model of enzymes, the enzyme’s inherent structure itself may also change when it is bound to a ligand. Thus, it is more beneficial to analyze the holo-structure of a protein as opposed to its apo-structure. However, the holo-structure may not always be available.
Having the holo-structure of a protein is usually preferred in drug design, when predicting protein binding sites and finding their shape/size. The holo-structure of a protein (conjugated protein) is the apo-protein combined with its prosthetic group (a ligand). Many enzymes may require cofactors or coenzymes in order to function fully. Such cofactors or coenzymes would be present within the holo-structure of the protein. Due to the induced fit model of enzymes, the enzyme’s inherent structure itself may also change when it is bound to a ligand. Thus, it is more beneficial to analyze the holo-structure of a protein as opposed to its apo-structure. However, the holo-structure may not always be available.
|
|
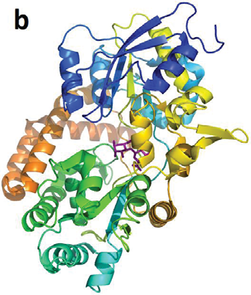
A protein’s (a) apo-structure (PDB: 1Y3Q) and (b) holo-structure (PDB: 1Y3N). It can be seen from the images that, in the holo-structure, the binding ligand (in purple stick representation) caused the protein to collapse more upon itself. After such a collapse, an existing cavity may be more “well defined” than it was in the apo-structure, which implies that detecting the cavity from the holo-structure would be easier than from the apo-structure.
B-Factor
The B-factor or the temperature factor is considered as an indication of the fluctuation of an atom in a protein. Atoms with low B-factors belong to the well-ordered part of the structure, whereas a large B-factor suggests high mobility of an atom. For interacting proteins, the interface residues are less flexible than the rest of the protein surface and, therefore, are often associated with small B-factors. Deeply buried atoms in the core of the protein are usually rigid with a low B-factor, and interfacial residues in protein binding complexes also have lower B-factors in comparison to the rest of the tertiary structural surface. The B-factor for each atom of a protein is available as part of the PDB (Protein Data Bank) file format.
The B-factor or the temperature factor is considered as an indication of the fluctuation of an atom in a protein. Atoms with low B-factors belong to the well-ordered part of the structure, whereas a large B-factor suggests high mobility of an atom. For interacting proteins, the interface residues are less flexible than the rest of the protein surface and, therefore, are often associated with small B-factors. Deeply buried atoms in the core of the protein are usually rigid with a low B-factor, and interfacial residues in protein binding complexes also have lower B-factors in comparison to the rest of the tertiary structural surface. The B-factor for each atom of a protein is available as part of the PDB (Protein Data Bank) file format.
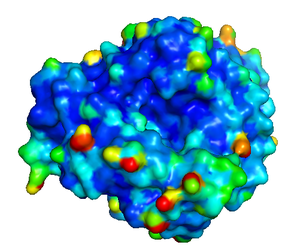
Salmonella typhimurium LT2 neuraminidase or STNA (2SIL) coloured by B-factor indicating vibrational movement (blue-red corresponds to low-high vibrational movement).
Van der Waals Interaction Energies
The interaction energy between a protein and a van der Waals probe can be used to locate energetically favourable binding sites. For example, in Q-SiteFinder, energetically favourable probe sites are clustered according to their spatial proximity and clusters are then ranked according to the sum of interaction energies for sites within each cluster. Specifically, it uses a methyl probe (−CH3) to calculate these interaction energies. Ligands have been found to bind to sites where the interaction energy within the protein is minimal, representing a stable binding site.
The interaction energy between a protein and a van der Waals probe can be used to locate energetically favourable binding sites. For example, in Q-SiteFinder, energetically favourable probe sites are clustered according to their spatial proximity and clusters are then ranked according to the sum of interaction energies for sites within each cluster. Specifically, it uses a methyl probe (−CH3) to calculate these interaction energies. Ligands have been found to bind to sites where the interaction energy within the protein is minimal, representing a stable binding site.
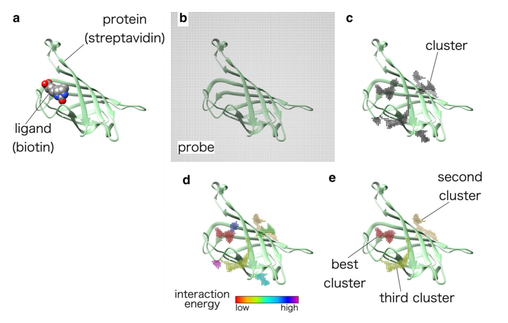
An example showing an energy-based approach to predicting protein binding sites.
Protein Dynamics
Proteins are flexible structures that can have more than one possible conformation, and this is why one static structure is not always enough to predict the binding sites. It is possible to produce ensembles of conformations using well-known molecular dynamics tools (e.g. GROMACS—GROningen MAchine for Chemical Simulations). Having such an ensemble of conformations, a binding site prediction program can then be used to predict “transient” and “conservative” pockets e.g. TRAPP (TRAnsient Pockets in Proteins).
Proteins are flexible structures that can have more than one possible conformation, and this is why one static structure is not always enough to predict the binding sites. It is possible to produce ensembles of conformations using well-known molecular dynamics tools (e.g. GROMACS—GROningen MAchine for Chemical Simulations). Having such an ensemble of conformations, a binding site prediction program can then be used to predict “transient” and “conservative” pockets e.g. TRAPP (TRAnsient Pockets in Proteins).
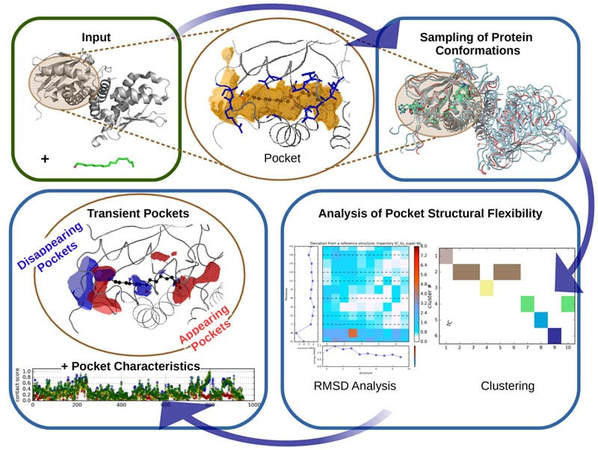
TRAPP workflow (note: RMSD analysis stands for root-mean-square deviation analysis of atomic positions which is the average distance between atoms of superimposed proteins).
Charge of Residue
The charge of a specific residue may affect its binding affinity, depending on the charge of the ligand it is binding to. For example, positively charged residues, such as arginine, are more likely to interact with the negatively charged backbone of DNA.
The charge of a specific residue may affect its binding affinity, depending on the charge of the ligand it is binding to. For example, positively charged residues, such as arginine, are more likely to interact with the negatively charged backbone of DNA.
